
Summary of GBS/CIDP Grants to Date
GBS|CIDP Research Report [PDF]
2004
Prof K.A. Sheikh, MD Johns Hopkins, Baltimore, MD
Determination of anti-ganglioside antibody affinity in patients with GBS
Antibody affinity and nerve injury in GBS is a complex issue. Our work points out at least 3 issues:
- Anti-ganglioside antibody affinity/avidity is likely not related to selective targeting, i.e., sensory vs motor nerve fiber recognition. Fine specificity of the antibodies is more relevant to this aspect. We had to clone a new anti-GD1a antibody with an order of magnitude higher affinity to make this point and also tested AMAN sera in this context. This was published in Brain 2008. See attached articles. GBS/CIDP foundation support acknowledged.
- Antibody affinity/avidity seems to be a factor in nerve fiber injury. This was shown with different GM1 ligands with low to extremely high affinities. In this study a monoclonal antibody with low affinity did not cause nerve injury, GBS patient anti-GM1 antibodies with moderate affinities produced nerve injury, and cholera toxin subunit B (ligand with highest affinity for GM1) produced more severe injury. This was published in J Neuroscience 2010. See attached articles. GBS/CIDP foundation support acknowledged.
- We also have unpublished data, which show that GBS patients have a range of affinities but the majority are in the moderate range. The antibody affinities are not as high as for example in M G. The caveat with the data are that these are cross-sectional data on antibodies in circulation. We have reason to believe that higher affinity antibodies may have shorter circulating half-lives. In order to produce injury moderate affinity antibodies must have robust Fc effector functions. We have preliminary data that indicate that Fc functions of anti-ganglioside antibodies are critical in producing nerve inflammation and injury.
Prof B. Arnason, MD University of Chicago, Chicago, IL
A New Approach to Treatment of the Guillain-Barre Syndrome
The basic idea was that we could use a construct we had developed with interesting immunomodulatory properties to treat EAN. It did not work: we still don’t quite understand why it didn’t. It didn’t work in EAE either even while it did work in an animal model for ITP. The construct is comprised of multiple copies of the CH2 region of IgG arranged linearly with the cysteines replaced by lysines to assure an open configuration so that the construct cross links multiple copies of all 3 classes of Fc gamma receptors, both activating and inhibiting, expressed on macrophages and thereby induce an off signal for cytokine secretion. Although this particular experiment failed other attempts, in addition to the ITP study, did not. When we put an antigenic peptide on the construct, the peptide was preferentially taken up by macrophages and dendritic cells with 10-100 times the antibody production seen when antigen alone was given. We now have US, Japanese, European and Australian patents for our technology, a start-up company called Iterative Therapeutics, a wet lab in an industrial complex across town, and 2 Small Business grants (SBIRs) from the NIH. One is to use our construct to augment the efficacy of Herceptin, since our construct really activates NK cells, perhaps because they express only activating receptors, and the other is to develop a vaccine to Botulinum toxin, a vaccine of potential interest to militaries concerned about bioterrorism. Another company has licensed our technology for an anthrax vaccine that they are developing. The funded projects, should they work out, will provide proof of principle, or so we hope, since if it works in one cancer it might well work in others, and ditto for vaccines. Thus, though the EAN project failed, the concept is still alive.
Prof. Hugh Willison Institute of Neurological Sciences, Glasgow, Scotland
Synthetic disialylgalactose immunoadsorbents deplete anti-GQ1b antibodies from autoimmune neuropathy sera
Acute and chronic autoimmune neuropathies, including Guillain-Barré syndromes (GBS) are often characterized by the presence of autoantibodies that react with neural gangliosides. Evidence from human and animal studies indicates that anti-ganglioside antibodies play a primary neuropathogenic role, and their rapid elimination from the circulation through specific immunoadsorption therapy thus has the potential to ameliorate the course of the disease. Here we have tested this therapeutic principle in the Miller Fisher variant of GBS that is associated serologically with acute phase anti-GQ1b ganglioside immunoglobulin G (IgG) antibodies, and in chronic ataxic neuropathies associated with persistently elevated immunoglobulin M (IgM) antibodies that react with GQ1b, GD3 and other disialylated gangliosides. Human and mouse anti-GQ1b IgG and IgM antibodies may also react with GD3, suggesting the shared terminal disialoside epitope could be involved in antibody binding. We thus synthesized the terminal trisaccharide, NeuAc(alpha2-8)NeuAc(alpha2-3)Gal common to GQ1b and GD3, and conjugated it to bovine serum albumin (BSA). This disialylgalactose glycoconjugate (DSG-BSA) binds anti-GQ1b antibodies in 32/58 (55%) human sera containing IgG or IgM anti-GQ1b antibodies at titres up to 1/130 000; it also binds a wide range of mouse monoclonal anti-GQ1b and -GD3 antibodies. When conjugated to Sepharose as mock therapeutic immmunoaffinity columns, the immobilized trisaccharide (DSG-Sepharose) eliminates anti-GQ1b antibodies from positive sera in proportion to their level of binding to DSG-BSA. Oligosaccharide-specific immunoadsorption therapy thus provides a new therapeutic approach to anti-GQ1b antibody-associated syndromes that could be applied to clinical practice. Furthermore, modification of the immobilized oligosaccharide epitopes to incorporate other glycan structures may allow this approach to be adapted to other forms of autoimmune neuropathy associated with uniform anti-glycolipid antibody profiles.
2005
Dr. M.N. Rasband University of Connecticut Health Center, Farmington, CT
The Role of Schwann Cell Microvilli at Nodes of Ranvier and in Guillain-Barre Syndrome
Autoantibodies to gangliosides such as GM1 have been proposed to disrupt nodes of Ranvier in peripheral motor nerve fibers and lead to Guillain-Barré syndrome, an autoimmune neuropathy characterized by acute limb weakness. At the nodes in myelinated axons, voltage-gated Na+ channels are highly concentrated and facilitate rapid action potential conduction. Using a disease model caused by immunization with gangliosides, we showed that Na+ channel clusters were disrupted or disappeared at abnormally lengthened nodes concomitant with deposition of IgG and complement products at the acute phase with progressing limb weakness (Susuki et al. J Neurosci 2007). Paranodal axoglial junctions, the nodal axonal cytoskeleton, and Schwann cell microvilli, all of which stabilize Na+ channel clusters, were also disrupted. During recovery, complement deposition at nodes decreased, and Na+ channels redistributed on both sides of affected nodes. These results suggest that Na+ channel alterations occur as a consequence of complement-mediated disruption of interactions between axons and Schwann cells. Our findings support the idea that the axonal form of Guillain-Barré syndrome is a disease that specifically disrupts the nodes of Ranvier. We also showed that gangliosides themselves are highly accumulated at and near nodes, and contribute to the formation and maintenance of the nodal Na+ channel clusters. In the mutant mice lacking gangliosides including GM1, ion channel clusters and structures at and near nodes were remarkably disrupted (Susuki et al. Glia 2007). Abnormal molecular organization at paranodes became more prominent with age. Furthermore, the defects were more prevalent in ventral than dorsal roots, and less frequent in mutant mice lacking the b-series gangliosides but with excess GM1. These results indicate that gangliosides contribute to stability and maintenance of nodes of Ranvier, and further support the critical roles of anti-ganglioside antibodies in Guillain-Barré syndrome.
Publications supported by this grant
Susuki K, Rasband MN, Tohyama K, Koibuchi K, Okamoto S, Funakoshi K, Hirata K, Baba H, Yuki N. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci 2007;27:3956-67
Susuki K, Baba H, Tohyama K, Kanai K, Kuwabara S, Hirata K, Furukawa K, Furukawa K, Rasband MN, Yuki N. Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia 2007;55:746-57
2005 – 2007
Prof R.A.C. Hughes Dept. of Clinical Neuroscience – Kings College London, England
A Pilot Randomized controlled trial of Methotrexate for Chronic Inflammatory Demyelinating Polyradiculoneuropathy RMC Trial
The GBS/CIDP Foundation grant funded the largest trial of an immunosuppressive agent for CIDP yet performed. The results were published in the prestigious international journal Lancet Neurology [RMC Trial Group 2009]. The trial was a high quality randomized study undertaken in 25 centers in 5 European countries. It compared methotrexate with identical appearing dummy or placebo tablets in people with CIDP already taking intravenous immunoglobulin or corticosteroids. After about 16 weeks, the dose of corticosteroids or intravenous immunoglobulin was decreased by 20% every 4 weeks provided that the participants did not worsen. The primary outcome was achieving a greater than 20% reduction in the dose of corticosteroids or intravenous immunoglobulin in the last 4 weeks of the trial compared with the first 4 weeks. Fifty-nine people completed the trial. Fourteen (52%) of the 27 taking methotrexate were able to reduce their corticosteroid or intravenous immunoglobulin dose but, unexpectedly, so were 14 (44%) of the 32 taking the dummy tablets. Thus the trial did not show any benefit from taking methotrexate but it did show, against expectations, that 44% were taking more corticosteroids or intravenous immunoglobulin than they needed. This has affected the design of subsequent CIDP treatment trials and had a major effect on the management of CIDP worldwide. It has led to the recommendation of drug holidays to check the need for continued treatment.
The grant paid for the employment of Dr. Mohamed Mahdi-Rogers, a neurologist originally from Sierra Leone, and allowed him to complete his training in London, UK, and secure a position as a Neuromuscular Disease Fellow. Thus the grant added to the small army of doctors qualified to look after people with CIDP. During his fellowship Dr. Mahdi-Rogers also completed an epidemiological study and a cost study of CIDP in south-east England [Mahdi-Rogers and Hughes 2013] and a Cochrane review of immunosuppressive treatment for CIDP [Mahdi-Rogers et al. 2013]. Support from the GBS/CIDP Foundation International has been acknowledged in all these publications.
Mahdi-Rogers M, Hughes RAC. Epidemiology of chronic inflammatory neuropathies in south east England. European Journal of Neurology 2013; [Epub ahead of print] PubMed PMID: 23679015.
Mahdi-Rogers M, van Doorn PA, Hughes RAC. Immunomodulatory treatment other than corticosteroids, immunoglobulin and plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database of Systematic Reviews 2013; CD003280.
RMC Trial Group. Randomized controlled trial of methotrexate for chronic inflammatory demyelinating polyradiculoneuropathy (RMC trial): a pilot, multicentre study. Lancet Neurol 2009; 8: 158-164.
2006
Prof K.A. Sheikh Johns Hopkins, Baltimore, MD
Reversal of anti-ganglioside antibody mediated inhibition of nerve regeneration with erythropoietin
Guillain-Barre´ syndrome (GBS) is a monophasic immune neuropathic disorder in which a significant proportion of patients have incomplete recovery. The patients with incomplete recovery almost always have some degree of failure of axon regeneration and target reinnervation. Anti-ganglioside antibodies (Abs) are the most commonly recognized autoimmune markers in all forms of GBS and specific Abs are associated with the slow/poor recovery. We recently demonstrated that specific anti-ganglioside Abs inhibit axonal regeneration and nerve repair in preclinical models by activation of small GTPase RhoA and its downstream effectors. The objective of this study was to determine whether erythropoietin (EPO), a pleiotropic cytokine with neuroprotective and neurotrophic properties, enhances nerve regeneration in preclinical cell culture and animal models of autoimmune neuropathy/nerve repair generated with monoclonal and patient derived Abs. Primary neuronal cultures and a standardized sciatic crush nerve model were used to assess the efficacy of EPO in reversing inhibitory effects of anti-ganglioside Abs on nerve repair. We found that EPO completely reversed the inhibitory effects of anti-ganglioside Abs on axon regeneration in cell culture models and significantly improved nerve regeneration/repair in an animal model. Moreover, EPO-induced proregenerative effects in nerve cells are through EPO receptors and Janus kinase 2. Signal transducer and activator of transcription 5 pathway and not via early direct modulation of small GTPase RhoA. These preclinical studies indicate that EPO is a viable candidate drug to develop further for neuroprotection and enhancing nerve repair in patients with GBS.
Prof U. Dianzani University of Eastern Piedmont, Novara, Italy
Search for biological markers of inflammatory demyelinating polyneuropathies development and progression: analysis of patients displaying defective function of the Fas apoptotic pathway
Is Defective Fas-Mediated T-Cell Apoptosis a Pathogenetic Mechanism in CIDP?
Starting from the concept that the immune response in the peripheral nervous system is shut
down through T-cell apoptosis, we aimed to assess whether defective T-cell apoptosis had a role in chronic inflammatory demyelinating polyneuropathy (CIDP) as shown for other autoimmune diseases, such as MS (Comi et al., 2000). We studied the Fas apoptotic pathway in patients with CIDP, Guillain-Barré syndrome (GBS), and in healthy controls, finding that CIDP patients displayed significantly higher T-cell survival to Fas-mediated apoptosis than both controls and GBS patients, whereas GBS patients and controls showed no difference. Moreover, the percentage of Fas-resistant (Fas-r) subjects, defined by the survival of more than 82% of their T cells to Fas-mediated apoptosis, was 52% in CIDP patients, 2% in controls, and 0% in GBS patients (Comi et al., 2006). The reason why we compared CIDP and GBS patients lies in the self-limiting nature of GBS as opposed to the persistent immune system activation of CIDP. Our hypothesis was that T-cell apoptosis might be a factor influencing, when effective, the self-limitation of the inflammatory process that we observed in GBS, or when defective, the disease persistence typical of CIDP. This concept was later reinforced by the finding that patients with acute-onset CIDP (a-CIDP) can be distinguished very early in the disease from patients with GBS by showing defective T-cell apoptosis. To show that, we used a twofold approach: initially, we compared Fas function in retrospective patients analyzed after diagnosis of GBS or a-CIDP and subsequently performed a prospective study analyzing Fas function at the onset of an acute inflammatory polyneuropathy and following up patients for the development of GBS or a-CIDP for at least 6 months. In both cases, we found that a-CIDP patients displayed significantly higher T-cell survival to Fas mediated apoptosis than controls and GBS patients. Moreover, single-patient analysis showed that all a-CIDP patients were Fas-r compared with one control and no GBS patients (Comi et al., 2009). One important question was whether Fas function may vary during the disease course. To clarify that, we tested the stability of Fas-mediated apoptosis with a second test at a time frame of about 1 year and found that variability was always <5%. We next tested whether the Fas pathway impairment had an inherited component, as was previously found in MS (Comi et al., 2000). We evaluated Fas-mediated T-cell death in families of Fas-r CIDP patients and found that in each family there was at least one Fasr parent (Comi et al., 2006). In previous studies, we showed that Fas-r patients with ALPS or MS produce molecules exerting a dominant-negative effect on Fas function (Comi et al., 2000; Ramenghi et al., 2000; Dianzani et al., 2003). To evaluate whether these molecules were also detectable in CIDP, we fused activated CD4+ T cells derived from Fas-r patients with the Fas-s HUT78 T-cell lines and cultured hybrid cells under the selective pressure of anti-Fas monoclonal antibodies (MAbs). Fusions from the Fas-r patients gave rise to Fas-r hybrid cell lines, whereas fusion from Fas-s control subjects did not (Comi et al., 2006).
Studying Fas-mediated cell death in CIDP patients with different clinical courses, we found that patients with a chronic progressive (CP) course had a significantly higher cell survival than patients with an RR course. Indeed, 70% of CP patients were Fas-r compared to only 20% of RR patients. After dividing CIDP patients according to neurophysiologic features at diagnosis, we found that patients with secondary axonal pattern had a significantly higher cell survival than those with a pure demyelinating form. All patients from the former group were Fas-r compared with only 24% of those from the latter (Comi et al., 2006). Finally, to better characterize the apoptosis defect, we evaluated the function of the extrinsic and intrinsic pathways of apoptosis by assessing the activation of caspase-8 induced by Fas triggering, and caspase-9 induced by etoposide, selectively acting on mitochondria in T-cell lines from Fas-r CIDP patients and Fas-s controls. Results showed that caspase-8 activity was significantly lower in CIDP patients than in controls, whereas no difference was found in caspase-9 activity. To assess whether defective caspase-8 activation was ascribable to increased expression of the FLIP inhibitor, we evaluated FLIP expression by Western blotting in the T-cell lines obtained from the same group of patients. Results showed no difference between patients and controls (Comi et al., 2006).
Our studies on T-cell apoptosis show that defective Fas function is a predisposing factor for CIDP development. Moreover, we provide evidence that T-cell Fas function may be a marker for identifying patients with an aggressive course and, in the case of acute onset, for discriminating between patients with a self-limiting disease and patients prone to develop a chronic course.
2007
Prof B. Soliven The University of Chicago, Chicago, IL
Spontaneous Autoimmune Polyneuropathy in the NOD Mouse
The antigen/s and the triggering mechanisms causing chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) remain elusive. B7-2 is one of the molecules expressed by antigen presenting cells and is required for optimal activation of T lymphocytes. When B7-2 is eliminated in non-obese diabetic (NOD) mice, animals do not develop diabetes, but instead they develop a spontaneous autoimmune polyneuropathy (SAP), which mimics CIDP in many ways.
Funding support from the GBS/CIDP Foundation International has facilitated our efforts to identify potential target antigens in B7-2 knockout NOD mice using a combination of immunologic methods that include proliferation assay, cytokine secretion, adoptive transfer and intravenous tolerance studies. We found the myelin P0 is one of the auto-antigens involved in the development of autoimmune neuropathy in this model, and that this protein is also expressed by Schwann cells in the pancreas. These findings suggest that a subset of patients with CIDP, particularly those with increased susceptibility to develop type 1 diabetes, may be mediated by autoimmunity against myelin P0. Data from this pilot grant were included in a manuscript published in the Journal of Immunology (Kim et al., 2008), and formed the basis of a successful NIH grant application.
Prof K.A. Sheikh Johns Hopkins, Baltimore, MD
Neuroprotection and Enhancement of Nerve Repair with Erythropoietin in EAN
Erythropoietin (EPO) is a pleiotropic cytokine originally identified for its role in erythropoiesis. In addition, in various preclinical models EPO exhibited protective activity against tissue injury. There is an urgent need for potent treatments of autoimmune driven disorders of the peripheral nervous system (PNS), such as the Guillain-Barre´ syndrome (GBS), a disabling autoimmune disease associated with relevant morbidity and mortality. To test the therapeutic potential of EPO in experimental autoimmune neuritis (EAN) – an animal model of human GBS – immunological and clinical effects were investigated in a preventive and a therapeutic paradigm. Treatment with EPO reduced clinical disease severity and if given therapeutically also shortened the recovery phase of EAN. Clinical findings were mirrored by decreased inflammation within the peripheral nerve, and myelin was well maintained in treated animals. In contrast, EPO increased the number of macrophages especially in later stages of the experimental disease phase. Furthermore, the anti-inflammatory cytokine transforming growth factor (TGF)-beta was upregulated in the treated cohorts. In vitro experiments revealed less proliferation of T cells in the presence of EPO and TGF-beta was moderately induced, while the secretion of other cytokines was almost not altered by EPO. Our data suggest that EPO revealed its beneficial properties by the induction of beneficial macrophages and the modulation of the immune system towards anti-inflammatory responses in the PNS. Further studies are warranted to elaborate the clinical usefulness of EPO for treating immune-mediated neuropathies in affected patients.
Prof R.P. Lisak Wayne State University, Detroit, MI
Identifying Targets of Tumor Necrosis Factor Alpha (TNF-a) Signaling that Inhibit Schwann Cell (SC) Differentiation
We previously showed that some inflammatory cytokines, primarily interferon–gamma (IFN-‐g) and tumor necrosis factor-‐alpha (TNF-‐a), which are found in the peripheral nerve of patients with GBS and CIDP, as well as in several animal models of GBS, are able to inhibit Schwann cells from expressing glycolipids associated with myelination in vitro. As part of our ongoing investigation into the relationships between inflammatory cytokines and Schwann cell biology, we have looked at the effects of both of these cytokines on Schwann cells. We are currently analyzing a very large set of data using gene array technology to determine if TNF-‐a alters the pattern of gene expression induced by cAMP, which simulates many of the effects of axolemma on Schwann cells in vitro. The gene array studies might provide a gene discovery approach to identify potential targets for decreasing or reversing inflammation-‐induced nerve demyelination. Although many laboratories, including our own, have shown that Schwann cells can upregulate major histocompatibility antigens (MHC) in vitro in response to inflammatory cytokines, in clinical and experimental inflammatory neuropathies, as well as in other diseases of the PNS, there is controversy over whether Schwann cells can upregulate major histocompatibility antigens or whether MHC expression is limited to infiltrating inflammatory cells. We postulated that Schwann cells in culture that are expressing myelin associated molecules, as many of them would be in a mature myelinated nerve, might be resistant to upregulation of MHC class II molecules as well as the adhesion molecule, intercellular adhesion molecule -‐1 (ICAM-‐1), also upregulated by cytokines. When we incubated undifferentiated Schwann cells with IFN‐g, the most potent stimulator of MHC Class II, both MHC Class II and ICAM-‐1were up-regulated in many of the cells. However, when Schwann cells were treated with cAMP in the presence or absence of IFN‐g, we found that when Schwann cells were stimulated by cAMP to differentiate and express myelin associated molecules, they did not express MHC Class II or ICAM-‐1. This indicates that the state of differentiation and perhaps myelination influences the response of SC to pro-stimulatory cytokines.
Dr. E.E. Ubogu Baylor College of Medicine, Houston, TX
CXCR3 Blockade and Inflammatory Demyelinating Polyradiculoneuropathies (IDP)
Funds from this award, in addition to funds received from the Baylor College of Medicine New Investigator Start-Up program supported the isolation, purification and characterization of primary human endoneurial endothelial cells from sciatic nerves (1). These cells are responsible for forming the restrictive blood-nerve barrier. This work has led to the development of an in vitro blood-nerve barrier system to study molecular determinants and signaling pathways potentially relevant to pathologic leukocyte trafficking in GBS and CIDP. Using this model, we demonstrated the effect of physiological pro-inflammatory cytokine stimulus at the blood-nerve barrier and showed the importance of alpha M integrin and intercellular adhesion molecule-1 [ICAM-1] (2), providing a framework to better understand pathologic leukocyte trafficking potentially relevant to targeted therapies for AIDP. Recent preliminary data using a reliable mouse model of AIDP called severe murine experimental autoimmune neuritis (sm-EAN) shows that CXCR3 blockade using a proprietary drug called AMG487 improves the clinical manifestations of sm-EAN when giving after disease onset (3), suggesting that drugs that block specific chemokine receptors may be useful future therapy for peripheral nerve inflammatory disorders such as AIDP.
2008
Dr. I.S.J. Merkies Universiteit Maastricht, The Netherlands
Peripheral Neuropathy Outcome Measures Standardization (PeriNom) Study
Different methods have been used to study patients with inflammatory polyneuropathies. The use of proper outcome measures is very important to facilitate adequate follow-up and evaluate treatment effects. The PeriNomS study aimed to expand the clinimetric knowledge on outcome measures at selected levels of outcome (mainly at the impairment, activity & participation, and quality of life levels) in patients with Guillain-Barré Syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), multifocal motor neuropathy (MMN) and monoclonal gammopathy of undetermined significance related polyneuropathy (MGUSP).
The PeriNomS study consisted of two parts:
The cross-sectional part focusing on comparative validity and reliability of the outcome measures, in which 122 clinically stable patients participated.
The longitudinal part was performed to obtain responsiveness data that may help to differentiate between comparable valid and reliable measures. The longitudinal part was an international, multi-centre collaborative approach of 26 neuromuscular centres. Newly diagnosed patients and patients with a relapse of their polyneuropathy (n=163) have been included and were examined 3 times (T0, T3, T12 months) in MMN (n=26) and MGUSP (n=23) or 5 times (T0, T1, T3, T6, and T12 months) in GBS (n=55) and CIDP (n=59). The inclusion period ended on January 1st, 2013, the dbase was closed and the main data at the impairment and activity and participation were presented at the 196th workshop, held in Naarden, the Netherlands under the auspice of the European NeuroMuscular Centre (ENMC). The ultimate goal was to get international consensus on a specific core set of outcome measures to be used in future clinical trials and follow-up studies in patients with inflammatory polyneuropathy, particularly focusing on GBS, CIDP, MGUSP, and MMN. At the 196th ENMC workshop, consensus was met for most illnesses at the various levels of assessing outcome. A paper on this will be published soon and at the PNS 2013 a presentation will be given on this topic.
Timeframe: the project was completed and all targets of inclusion were met, for both cross-sectional and longitudinal recruitments.
Prof P.A. van Doorn Erasmus MC, The Netherlands
Autonomic Failure and pain in Guillain-Barre Syndrome: A skin biopsy study
We performed a prospective study in 32 patients with Guillain–Barré syndrome (GBS) or its variants to correlate intraepidermal nerve fiber density (IENFD) at the distal leg and lumbar region with pain, autonomic dysfunction, and outcome. In the acute phase, IENFD was reduced in 60% and 61.9% of patients at the distal leg and lumbar region, respectively. In the acute phase, 43.7% of patients complained of neuropathic pain. Their IENFD at the distal leg was significantly lower than in patients without pain (P < .001) and correlated with pain intensity (rs = [1]0.51; P = .003). Intriguingly, also patients with the pure motor variant of GBS and pain had low IENFD. At 6-month follow-up, only 3 patients complained of persisting neuropathic pain, whereas 3 patients reported late-onset pain symptoms. IENFD in the acute phase did not predict presence or intensity of pain at 6-month follow-up. IENFD in the acute phase did not correlate with clinical dysautonomia or GBS severity at nadir. However, it correlated with poorer GBS disability score at 6 months (P = .04), GBS score at nadir (P = .03), and clinically probable dysautonomia (P = .004). At 6-month follow-up, median IENFD remained significantly low both at the distal leg (P = .024) and lumbar region (P = .005). Double and triple staining confocal microscope studies showed diffuse damage of myelinated dermal nerves along with axonal degeneration, and mononuclear cell infiltration. Unmyelinated and myelinated skin nerves are diffusely affected in GBS and its variants, including the pure motor form. IENFD declines early, remains low over time, correlates with pain severity in the acute phase, and may predict long-term disability.
Prof R.A. Lewis Wayne State University. Detroit, MI
Database of Multifocal Motor Neuropathy and Other Inflammatory Neuropathies
The project was to create a registry/database of patients with Multifocal Motor Neuropathy and if successful to be utilized in the future for other inflammatory neuropathies. The funds were utilized to contract with the University of Maryland Bioinformatics Department to assist in the creation of the database, and the implementation of a web-based data entry system. A committee of neurologists active in the Inflammatory Neuropathy Consortium met at the INC meetings and dialogued by email to work on the information that would be put into the database.
A number of issues were identified that required attention before a product could be implemented. These included:
- HIPPA compliance and patient anonymity,
- Developing a method to make sure that patients actually had MMN,
- Keeping the amount of information that was required to a minimum but having enough information to be useful, and
- Getting physicians to use the registry/database.
It was decided that the initial product would be a registry that would include demographics, whether the patient met either AANEM or EFNS criteria, whether conduction block was identified, whether there were any sensory signs or symptoms and whether GM1 antibodies were present. In addition, information on treatments used and their efficacy was included.
The project took over 2 years, but the registry was developed and was initiated. However, full implementation stalled and is currently not active. The major problem that occurred was the dismantling of the University of Maryland Bioinfomatics Department. The registry is still available and there has been interest (Baxter remains interested) in working with Dr. Leonard van den Berg and others to reinvigorate the project. One approach that is very appealing is to utilize the mechanism created for IGOS to do a very similar project on MMN.
In summary, the project on creating an MMN registry and database was partially successful in that a product was created that can be utilized in the future. The difficulties in implementation were frustrating but the overall goal of developing a network of centers that can do future treatment trials in MMN is moving forward.
2009
Prof K.A. Sheikh University of Texas, Houston, TX
Engineering chimeric protein(s) to enhance nerve repair in antibody-mediated preclinical models of autoimmune neuropathy
We had funding for one year but spent at least 2 years on this project. Progress has been slow on this one and include generation of six different constructs of human and mouse EPO conjugated with 3 different mouse IgG Fc isotypes. We have tried to express mouse EPO constructs in a mammalian cell line and had only limited success with one construct. The level of protein expression is extremely low. Despite our best efforts, we have been able to generate sufficient chimeric protein to carry out pharmacokinetic and efficacy studies in animals. The project is currently dormant due to funding. But once funding is available we plan to have some of these chimeric proteins expressed in sf9 insect cells. I have talked with protein expression experts and they do not have any additional suggestions except trying the insect cells. Apparently, expressing chimeric proteins in mammalian systems is tedious and unpredictable hit or miss. If insect expression provides us sufficient chimeric protein then will test this in animal studies. I have asked the lab to prepare a power point containing key data showing protein generation, which is attached. All EPO-Fc constructs and transfected CHO cell lines are available as a resource for sharing.
Dr. B.C. Jacobs Erasmus MC, The Netherlands
Prognostic models for Guillain-Barré syndrome.
Guillain-Barré syndrome (GBS) has a highly variable clinical course and outcome. Despite the fact that various studies described prognostic factors in GBS, these are difficult to apply in clinical practice. To support patients, family and neurologists we have developed various validated prognostic models which are based on clinical characteristics which can be easily obtained at the bedside of the patient. These models were developed to predict in individual patients with GBS the chance of respiratory failure (Walgaard et al. Brain 2010) and severe disability at one to six months (Walgaard et al. Neurology 2011). These models have found their way to the clinic and are for example used in the Dutch National Guideline for the treatment of GBS. The next step was to further improve and validate these prognostic models in a large cohort of GBS patients. Such numbers of patients can only be reached by large scale international collaboration. With support from the GBS-CIDP Foundation International we have initiated the International GBS Outcome Study (IGOS). IGOS is conducted by the members of the Inflammatory Neuropathy Consortium (INC). The aim of the IGOS is to identify the clinical and biological predictors of the clinical course and outcome of GBS. Supported by this grant a website and web based data entry system and database was developed by a professional web designer. Data are collected at 7 standard time points during a follow-up of one year, but there is an opportunity to expand the follow-up to 2 and 3 years. Using this system detailed and standardized data are obtained regarding the demography, preceding events, neurological deficits, diagnostic investigations, treatment , complications and outcome. Several validated outcome scores are used to describe in detail the most important consequences to the patients, including disability, pain and fatigue. After the prototype website was developed various extensive test rounds were conducted in participating centers from various countries to further improve the website and database. Data are collected in a coded form without reference to personal information and stored in an environment which is highly secured and protected. The website and data base is up and running and currently used by more than 100 research centers from North- and South-America, Europe, Asia and Australia participating in IGOS. The website is maintained daily and can be visited at www.gbsstudies.org. At this website a GBS tool is presented to predict the clinical course in individual patients.
Dr. H. C. Lehmann Heinrich-Heine University, Germany
Schwann cell replacement therapy for chronically denervated nerves
Our project is based on the well-recognized clinical dilemma that patients with Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy (CIDP) often show an incomplete recovery and poor functional outcome after a prolonged, severe disease course.
Previous experimental studies suggested that incomplete recovery in peripheral nerve disorders is partially attributable to morphological changes in the distal segments of chronically denervated nerves. There is evidence that Schwann cells, which provide trophic support under normal conditions, become atrophic and lose their ability to support the regeneration of axons after prolonged denervation.
Thus we wanted to explore the underlying cellular mechanisms of this chronic denervation condition in GBS and CIDP. Moreover we pursue the idea that axonal regeneration within those chronic denervated nerve segments in patients with GBS and CIDP could be enhanced by transplantation of freshly extracted, healthy Schwann cells. To address this clinically important issue we adapted and modified an animal model, which mimics the situation of chronic denervation, comparable to those of GBS/CIDP patients after a prolonged disease course with secondary axonal loss. The project was started in 2010 and is still ongoing. The initial phase of this project was used to explore different experimental paradigms to address basic practical issues in this animal model such as timing, fate and application route of endoneurial Schwann cell transplantation. Our preliminary experiments suggest that the transplantation of Schwann cells into chronically denervated nerve stumps can enhance regeneration, provided that narrow temporal-spatial conditions are carefully taken into account. We also observed that grafted Schwann cells do not survive over prolonged time periods, which has implications for the primary mode of action by which those grafts enhance regeneration of injured axons (transient growth factor expression versus remyelination).
In ongoing experiments we are currently investigating if Schwann cells that are exposed to an inflammatory environment like in GBS and CIDP preserve their regeneration promoting phenotype and their utility to serve as cellular graft for this model. We anticipate that the preclinical data which are generated by our proof-of-principle study will provide important information how future cell-replacement studies have to be designed to achieve highest benefit for patients with GBS and CIDP.
2011
Dr. I Nachamkin University of Pennsylvania, Philadelphia, PA
Genome Wide Association Study of Guillain-Barre Syndrome
No work was completed on this project. Money has been returned.
Prof J. Devaux Research Center of Neurobiology-Neurophysiology of Marseille, Marseille, France
The role of antibodies against proteins of the node of Ranvier in the pathogenesis of Guillain-Barre syndrome: finding novel immune targets.
Background Chronic inflammatory demyelinating polyneuropathy (CIDP) is a peripheral nervous system (PNS) disease with progressive or relapsing-remitting (RR) course1. An autoimmune attack against the PNS myelin, with the involvement of both the cellular and humoral components of the immune response is the main pathogenic event2. A central role is played by T helper (TH) cells secreting cytokines supporting macrophage and B cell activation2. Recent research has shift the interest from a traditional view of an imbalance between pro-inflammatory TH1 and anti-inflammatory TH2 cells as the main mechanism of autoimmunity, to a more complex scenario where TH17 cells control the pro-inflammatory component, modulating also the activity of TH1 cells3. Most data on the TH17 role have been obtained in common autoimmune diseases, including multiple sclerosis (MS), but their role has also been suggested in CIDP since TH17 cells and IL-17 levels have been found to be increased in active CIDP4 in the peripheral blood.
Osteopontin (OPN) is a cytokine that may influence development of autoimmune diseases through its capacity to enhance both TH1 and TH17 cell responses5,6. High OPN levels have been detected in several autoimmune diseases, including EAE and MS, especially during relapses5. Moreover, single nucleotide polymorphisms (SNPs) of the OPN gene have been associated with several autoimmune diseases7-9, and a link has been suggested with the increased protein levels12. In particular, we detected four SNPs (+282T>C in exon VI;+750C>T in exon VII; +1083A>G and +1239A>C in 3′ UTR) of the OPN gene which form three haplotype combinations (haplotype-A: 282T-750C-1083A-1239A; haplotype-B: 282C-750T-1083A-1239C; haplotype-C: 282C-750T-1083G-1239C) displaying associations with several autoimmune diseases; in fact, subjects carrying haplotype-B or -C displayed 1.5 higher risk of MS, type 1 diabetes, and systemic lupus erythematosus, and 8 fold higher risk of autoimmune lymphoproliferative syndrome (ALPS), than haplotype-A homozygotes. Several data suggested that these haplotypes influence OPN levels and, in particular, haplotype-B and -C are associated with higher levels of OPN possibly because their mRNAs was more stable than the haplotype-A mRNA9. The OPN role in MS is supported by the finding that OPN-deficient mice are resistant to progressive EAE and have frequent remissions10 ; in these mice, myelin-reactive T cells produce more IL-10 and less IFNγ than in wild type mice. A further point is that Steinman et al.11 showed that low levels of anti-OPN auto-antibodies (auto-Ab) were produced in EAE mice, and boosting their production by DNA vaccinations with a plasmid encoding OPN substantially ameliorated the chronic course of EAE. These findings are in line with reports suggesting that production of auto-Ab against pro-inflammatory cytokines plays a physiologic role in down modulation of the inflammatory response12.
Several studies have explored the role of OPN in autoimmune diseases, but little is known about its role in inflammatory polyneuropathies. A role is suggested by the finding that OPN is constitutively expressed in the PNS and up-regulated in rat experimental autoimmune neuritis13. It is noteworthy that OPN can inhibit T cell apoptosis14 that we previously showed to be defective in CIDP15.
In this context, this study may bridge the knowledge gap on the role of other apoptotic pathways which may be involved in controlling T cell survival in parallel to the direct pathway mediated by Fas.
Given these premises, the main objectives of this project were: a) to explore the role of OPN and anti-OPN auto-antibodies in CIDP; b) to further characterize apoptotic pathways involved in T cell survival in CIDP. Specific aims were the following: 1) To evaluate the levels of OPN and anti-OPN auto-antibodies in CIDP and GBS and assess their correlations with disease status (relapse, remission, chronic phase). 2) To type the OPN gene haplotypes and analyze other genes involved in T cell apoptosis in CIDP and GBS patients.
Patients and methods. We enrolled 62 patients with CIDP16, 30 with Guillain-Barré syndrome (GBS) and 50 age matched healthy controls. We assessed OPN plasma levels with a commercially available ELISA kit (IBL diagnostics). The presence of anti-OPN antibodies was demonstrated by western blot, whereas anti-OPN Ab levels were assessed with a custom made ELISA. Thereafter, we compared OPN plasma levels in patients CIDP with different clinical outcomes (progressive vs. relapsing course, axonal vs. demyelinating damage). Finally, we analyzed OPN gene haplotypes (+1239A>C), known to influence protein plasma levels, and variants of Perforin (PRF) gene that is involved in apoptosis and play a role several autoimmune diseases17.
Results. CIDP patients displayed significantly higher OPN plasma levels compared to both GBS patients and healthy controls (median value in ng/ml: 250 vs 165 and 160 respectively, p=0.015).
OPN plasma levels were higher in patients with progressive vs. relapsing course and secondary axonal damage vs. demyelination (median value in ng/ml: 270 vs. 165 and 240 vs. 170 respectively, p=0.023 and p=0.035). Moreover, we found that CIDP patients showed higher anti-OPN antibody plasma levels compared to controls (median titer 0.36 vs. 0.14, p=0.02). On the genetic side, OPN gene variants did not seem to be differently distributed among tested subjects (data not shown). On the contrary, PRF gene variations which are known to influence protein function, were differently distributed in patients compared to controls. A91V variations were present in 19% of patients versus 9% of controls (p>0.05). Moreover, we found two CIDP patients carrying new PRF mutations which were found to display a biologic activity.
Conclusion. This is the first report highlighting the role of OPN in CIDP. Increase of OPN plasma levels is associated to a chronic disease evolution, being absent in a self-limiting disease like GBS and more frequent in CIDP sub-forms with aggressive evolution. Moreover, patients with CIDP show higher anti-OPN Ab levels. A further step would imply the characterization of outcome in relation to both OPN and anti-OPN Ab levels. Neither protein nor antibody levels seem to be influenced by the genetic background since we found no difference in OPN SNPs between patients and controls. PFR gene variations/mutations may represent a predisposing factor for CIDP development.
Dr E.E. Ubogu Baylor College of Medicine, Houston, TX
CCL2-CCR2 blockade as targeted immune- modulatory therapy for inflammatory demyelinating polyradiculoneuropathies.
Funds from this award, in addition to funds received from the National Institutes of Health supported our work on the effects of functional inhibition of CCR2 signaling in immune-mediated demyelinating neuritis using the sm-EAN model. Preliminary data obtained following sm-EAN induction using a CCR2 knockout mouse showed that these mice were resistant to developing clinical manifestations of the disease, suggesting that CCR2 may be essential in the pathogenesis of acute demyelinating neuritis (3). Unpublished work (in preparation for submission) shows that a commercially available CCR2 inhibitor RS 102895 effectively treated mice with sm-EAN days after disease onset, with evidence of improved electrophysiologic parameters, and reduced inflammation and demyelination compared to untreated controls. Data from this study further suggests that targeted inhibition of specific chemokine receptors may be useful future therapies for AIDP and related disorders. Funds from the Foundation also supported the publication of a review article on chemokines in peripheral nervous system inflammation published in 2011(4).
Yosef N, Xia RH, Ubogu EE. Development and characterization of a novel human in vitro blood-nerve barrier model using primary endoneurial endothelial cells. Journal of Neuropathology and Experimental Neurology 2010; 69:82-97.
Yosef N, Ubogu EE. αMβ2-integrin-intercellular adhesion molecule-1 interactions drive the flow-dependent trafficking of Guillain-Barré syndrome patient derived mononuclear leukocytes at the blood-nerve barrier in vitro. Journal of Cellular Physiology 2012; 227:3857-3875.
Chiang S, Ubogu EE. The role of chemokines in Guillain-Barré syndrome. Muscle and Nerve 2013 (In press. On-line version: DOI: 10.1002/mus.23829, published February 28th, 2013).
Ubogu EE. Chemokine receptors as specific anti-inflammatory targets in peripheral nerves. Endocrine, Metabolic and Immune Disorders-Drug Targets 2011;11:141-153.
2011
Dr. C. Como University of Eastern Piedmont Amedeo Avogadro, Itally
The role of osteopontin and anti-osteopontin antibodies in CIDP
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a peripheral nervous system (PNS) disease with progressive or relapsing-remitting (RR) course1. An autoimmune attack against the PNS myelin, with the involvement of both the cellular and humoral components of the immune response is the main pathogenic event2. A central role is played by T helper (TH) cells secreting cytokines supporting macrophage and B cell activation2. Recent research has shift the interest from a traditional view of an imbalance between pro-inflammatory TH1 and anti-inflammatory TH2 cells as the main mechanism of autoimmunity, to a more complex scenario where TH17 cells control the pro-inflammatory component, modulating also the activity of TH1 cells3. Most data on the TH17 role have been obtained in common autoimmune diseases, including multiple sclerosis (MS), but their role has also been suggested in CIDP since TH17 cells and IL-17 levels have been found to be increased in active CIDP4 in the peripheral blood.
Osteopontin (OPN) is a cytokine that may influence development of autoimmune diseases through its capacity to enhance both TH1 and TH17 cell responses5,6. High OPN levels have been detected in several autoimmune diseases, including EAE and MS, especially during relapses5. Moreover, single nucleotide polymorphisms (SNPs) of the OPN gene have been associated with several autoimmune diseases7-9, and a link has been suggested with the increased protein levels12. In particular, we detected four SNPs (+282T>C in exon VI;+750C>T in exon VII; +1083A>G and +1239A>C in 3′ UTR) of the OPN gene which form three haplotype combinations (haplotype-A: 282T-750C-1083A-1239A; haplotype-B: 282C-750T-1083A-1239C; haplotype-C: 282C-750T-1083G-1239C) displaying associations with several autoimmune diseases; in fact, subjects carrying haplotype-B or -C displayed 1.5 higher risk of MS, type 1 diabetes, and systemic lupus erythematosus, and 8 fold higher risk of autoimmune lymphoproliferative syndrome (ALPS), than haplotype-A homozygotes. Several data suggested that these haplotypes influence OPN levels and, in particular, haplotype-B and -C are associated with higher levels of OPN possibly because their mRNAs was more stable than the haplotype-A mRNA9. The OPN role in MS is supported by the finding that OPN-deficient mice are resistant to progressive EAE and have frequent remissions10 ; in these mice, myelin-reactive T cells produce more IL-10 and less IFNγ than in wild type mice. A further point is that Steinman et al.11 showed that low levels of anti-OPN auto-antibodies (auto-Ab) were produced in EAE mice, and boosting their production by DNA vaccinations with a plasmid encoding OPN substantially ameliorated the chronic course of EAE. These findings are in line with reports suggesting that production of auto-Ab against pro-inflammatory cytokines plays a physiologic role in down modulation of the inflammatory response12.
Several studies have explored the role of OPN in autoimmune diseases, but little is known about its role in inflammatory polyneuropathies. A role is suggested by the finding that OPN is constitutively expressed in the PNS and up-regulated in rat experimental autoimmune neuritis13. It is noteworthy that OPN can inhibit T cell apoptosis14, that we previously showed to be defective in CIDP15.
In this context, this study may bridge the knowledge gap on the role of other apoptotic pathways which may be involved in controlling T cell survival in parallel to the direct pathway mediated by Fas.
Given these premises, the main objectives of this project were: a) to explore the role of OPN and anti-OPN auto-antibodies in CIDP; b) to further characterize apoptotic pathways involved in T cell survival in CIDP. Specific aims were the following: 1) To evaluate the levels of OPN and anti-OPN auto-antibodies in CIDP and GBS and assess their correlations with disease status (relapse, remission, chronic phase). 2) To type the OPN gene haplotypes and analyze other genes involved in T cell apoptosis in CIDP and GBS patients.
Patients and methods. We enrolled 62 patients with CIDP16, 30 with Guillain Barré syndrome (GBS) and 50 age matched healthy controls. We assessed OPN plasma levels with a commercially available ELISA kit (IBL diagnostics). The presence of anti-OPN antibodies was demonstrated by western blot, whereas anti-OPN Ab levels were assessed with a custom made ELISA. Thereafter, we compared OPN plasma levels in patients CIDP with different clinical outcomes (progressive vs relapsing course, axonal vs demyelinating damage). Finally, we analyzed OPN gene haplotypes (+1239A>C), known to influence protein plasma levels, and variants of Perforin (PRF) gene that is involved in apoptosis and play a role several autoimmune diseases17.
Results. CIDP patients displayed significantly higher OPN plasma levels compared to both GBS patients and healthy controls (median value in ng/ml: 250 vs 165 and 160 respectively, p=0.015).
OPN plasma levels were higher in patients with progressive vs relapsing course course and secondary axonal damage vs demyelination (median value in ng/ml: 270 vs 165 and 240 vs 170 respectively, p=0.023 and p=0.035).
Moreover, we found that CIDP patients showed higher anti-OPN antibody plasma levels compared to controls (median titer 0.36 vs 0.14, p=0.02).
On the genetic side, OPN gene variants did not seem to be differently distributed among tested subjects (data not shown). On the contrary, PRF gene variations which are known to influence protein function, were differently distributed in patients compared to controls. A91V variations were present in 19% of patients versus 9% of controls (p>0.05). Moreover, we found two CIDP patients carrying new PRF mutations which were found to display a biologic activity.
Conclusion. This is the first report highlighting the role of OPN in CIDP. Increase of OPN plasma levels is associated to a chronic disease evolution, being absent in a self-limiting disease like GBS and more frequent in CIDP sub-forms with aggressive evolution. Moreover, patients with CIDP show higher anti-OPN Ab levels. A further step would imply the characterization of outcome in relation to both OPN and anti-OPN Ab levels. Neither protein nor antibody levels seem to be influenced by the genetic background since we found no difference in OPN SNPs between patients and controls. PFR gene variations/mutations may represent a predisposing factor for CIDP development.
Dr G. Zhang The University of Texas Medical School of Houston, TX
Examining antagonistic/synergistic effects of IVIg and erythropoietin (EPO) in a model of autoimmune neuropathy
OBJECTIVE: To examine the potential antagonistic/synergistic effects of IVIg and EPO in preclinical models of anti-ganglioside antibody (Ab)-mediated neuropathy and inhibition of axon regeneration. BACKGROUND: Anti-ganglioside antibodies (Abs) are commonly present in patients with Guillain-Barré syndrome (GBS). We and others have shown that intravenous immunoglobulins (IVIg) can prevent anti-ganglioside Abs mediated complement dependent nerve cell/fiber injury. Our recent studies suggest that erythropoietin (EPO) enhances nerve repair in preclinical models of GBS. IVIg is now the standard treatment for GBS and a new trial to examine the efficacy of EPO in GBS would have to be done as add on therapy with IVIg. It is necessary to demonstrate that EPO and IVIg do not have antagonistic effects. It would also be important to know whether these two medications have synergistic effects to design an effective administration schedule in patients with GBS. DESIGH/METHODS: Previously published preclinical models of autoimmune neuropathy mediated by anti-ganlioside Ab were used to examine the potential antagonistic/synergistic effects of IVIg and EPO. RESULTS: Our results show that individual treatments with either IVIg or EPO provide significant protection, i.e., postpone the anti-ganglioside Ab- and complement-mediated neural injury to the neurites and neuronal cell body, in the neurocytotoxicity assays. Notably, no antagonistic effects were seen with the combined use of IVIg and EPO in these assays. Our data suggest that EPO and IVIg have synergistic effects in protecting neuronal cells from this anti-ganglioside Ab-mediated complement-dependent injury. CONCLUSTIONS: Our results support the conclusion that EPO can be useful add-on therapy to the current standard IVIg treatment for GBS. This novel combination therapy can be potentially beneficial for protecting nerve from injury during the acute phase and enhancing nerve repair in the recovery period in GBS patients.
Acknowledgements: These studies were supported by GBS/CIDP Foundation and NIH/NINDS (NS42888 and NS54962).
Zhang, G., Bogdanova, N., Song, J., Sheikh, K. Synergistic effects of IVIg and erythropoietin (EPO) in preclinical models of autoimmune neuropathy. Neurology 2013; 80: P01.154.
Dr B.C. Jacobs Erasmus MC, The Netherlands
Implementation of the International Guillain-Barre Syndrome Outcome Study (IGOS)
The International GBS Outcome Study (IGOS) is a worldwide prospective cohort study conducted by the International Neuropathy Consortium (INC) that aims to identify the clinical and biological determinants of disease progression and recovery in GBS. A website and database for inclusion of patients and data in this study was recently developed with support from the GBS-CIDP Foundation. We aim to include at least 1000 patients with GBS in IGOS. The grant was used to implement the study and we performed the following activities: (1) Neurologists from all over the world were approached to participate. At present (September 2013) more than 250 neurologists from 181 research centers from 18 countries are participating or preparing to participate. (2) The IGOS secretary supported these participants to obtain approval from the local Institutional Review Boards (IRB). The current state is that 106 centers have IRB approval. (3) Information sheet for patients were translated in 8 foreign languages by the participant and official translation agencies. (4) We facilitated the data entry and conducted regularly data quality controls. (5) Maintenance and updates of the IGOS website and database (see www.gbsstudies.org). (6) Support with registration, transport and storage of the collected biomaterials. (7) Two meetings for the IGOS consortium were organized, which were held during the INC congress in Rotterdam, The Netherlands in July 2012, and during the PNS congress in Saint-Malo, France in June 2013. IGOS officially started in July 2012. At present 208 GBS patients are included from 14 countries including the UK (53 patients), USA (41 patients), Italy (25 patients), Denmark (25 patients), Netherlands (24 patients), Germany (13 patients), Spain (12 patients), Japan (6 patients), France (3 patients), Argentina (3 patients), Canada (3 patients), Belgium (1 patient), Australia (1 patient), and Malaysia (1 patient). Researchers from Bangladesh, Brazil, India and Taiwan also have shown interest to participate and are in the process of acquiring IRB approval. IGOS will result in the world’s largest prospectively collected clinical database and biobank which will be used to understand the processes that determine disease cause, progression and recovery. These insights will form the basis to accurately predict the clinical course of individual patients with GBS and to develop new strategies to personalize therapy.
Dr. I.S.J. Merkies Universiteit Maastricht, The Netherlands
Peripheral Neuropathy Outcome Measures Standardization (PeriNoms) Study
See above and attached report
2012
Dr C.A. Massaad The University of Texas Health Science Center, Houston, TX
Comparing sialylated IgG and Fc fragments with IVIG in an anti-ganglioside antibody induced neuropathy model of GBS
High doses of Intravenous immunoglobulin (IVIG), requiring infusion over several days, are widely used for the treatment of Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Recent studies indicate that autoantibody- and Fcγ receptor-mediated inflammatory cascade, is abrogated by a sialic acid-enriched minor populations present in commercial IVIG preparations. We have recently established that Fcγ receptor activation is critically involved in neural injury in a mouse model of GBS. In this study, we investigated the effects of sialic acid-enriched IVIG (seIVIG) and compared them with whole/native IVIG (nIVIG) and sialic acid-deficient IVIG (sdIVIG) in an established model of nerve repair in which anti-ganglioside antibodies significantly inhibit axon regeneration. We used lectin affinity-chromatography to generate various IVIG fractions and used them in a passive transfer model of anti-ganglioside antibody-mediated inhibition of axon regeneration. Behavioral, electrophysiological, and morphological studies showed beneficial effects of nIVIG and seIVIG on abrogating the adverse effects of anti-ganglioside antibodies on nerve repair. Notably, seIVIG was equally efficacious at a dose tenfold lower than the nIVIG. sdIVIG was ineffective in modulating anti-glycan antibody-mediated inhibition of nerve repair. Further, our lectin chromatographic studies indicate that various commercial IVIG preparations have up to 2-fold variation in seIVIG content. Our findings have translational implications as smaller amounts of IVIG and shorter infusion times are preferable because common side effects of IVIG relate with rate of infusion and amount administered. Shorter infusion times are likely to increase patient acceptance of repeated IVIG infusions necessary for the treatment of chronic immune neuropathies such as CIDP.
Dr E.P. Simpson Methodist Neurological Institute, Houston, TX
CD4+CD25+ regulatory T cells as Potential Biomarkers of Pathogenesis and Response in Therapy in CIDP Patients
No work was completed on this project and the monies are being refunded.
Dr. L. Cousens EpiVax, Inc., Providence, RI
Tregitopes: A Novel Immunomodulatory Therapy for CIDP
Regulatory T cells (Treg) suppress inflammation and therefore are critically important for resolution of autoimmune chronic inflammatory demyelinating polyneuropathy (CIDP). CIDP treatment focuses on inhibiting inflammation, and thus demyelination and secondary axonal loss. IVIG, an approved CIDP therapy, induces expansion of Treg in a number of human diseases and animal models. The efficacy of IVIG has been demonstrated in clinical trials, although the precise mechanism of action by which IVIG exerts its immunomodulatory effects is not clearly understood. Alternative therapies are needed, as IVIG cost and availability remain major barriers to meeting the medical needs of CIDP patients. In recent work, EpiVax has identified one trigger for Treg expansion, natural T regulatory cell epitopes (Tregitopes) contained in immunoglobulin G, and therefore in IVIG. Tregitope peptides have now been shown to replicate the effects of IVIG in a number of in vitro and in vivo models. The IVIG-like effects of Tregitopes have been validated by independent laboratories in mouse models of Type-1 Diabetes, multiple sclerosis, transplant rejection, allergy and asthma, thus confirming our primary observations. We propose Tregitopes as a potential substitute for IVIG in the treatment of certain autoimmune diseases. We reason that CIDP will also respond to Tregitope immune-modulating therapy, and that this may prove to be a more specific and accessible therapy relative to IVIG. We are studying the therapeutic application of Tregitopes to ameliorate disease in the mouse model of CIDP disease. These studies will provide critical insight into basic mechanisms by which Tregitopes engage Treg to induce tolerance and will define key parameters for Tregitope therapy in CIDP.
2013 SYNOPSIS OF GRANT AWARDED AND THE REPORTED RESULTS
GBS in LOW INCOME COUNTRIES
Hubert P. Endtz, MD PhD1,2,*, Zhahir Islam, PhD1, MD1, Deen Mohammed, MD PhD3, Bart C. Jacobs, MD PhD4 ,1International Centre for Diarrheal Disease Research, Dhaka, Bangladesh;
2Medical Microbiology and Infectious Diseases, Erasmus MC, Rotterdam, The Netherlands;
3National Institute of Neuroscience, Dhaka, Bangladesh and Dhaka Medical College Hospital;
4Neurology and Immunology, Erasmus MC, Rotterdam, The Netherlands;
Objectives. The proposed project involves two objectives: (1) to define the clinical course and outcome of Guillain-Barré syndrome (GBS) in low-income countries and to identify the preceding infections and other factors that influence outcome, and (2) to investigate the safety of small volume plasma exchange (SVPE) in patients with GBS in Bangladesh. Methods. (1) Three hospitals in Dhaka, Bangladesh will participate in the International GBS Outcome Study (IGOS), which will provide a website and infrastructure for standardized measurement of outcome. (2) Twenty patients with severe GBS who are unable to walk will be included in a phase II study investigating the safety of small volume Plasma exchange, using the IGOS database for data registry
Impact: Currently there is no standard care and treatment for the Guillain-Barré syndrome (GBS) in low-income countries due to a lack of an available plasmapheresis facilities and high cost of Intravenous Immunoglobulin. This proposal will study the clinical course of GBS in Bangladesh in detail. They will also assess the safety of a new, low-cost treatment for patients with GBS from developing countries, who would otherwise be deprived of effective treatment. Participation in IGOS will provide a unique opportunity for researchers from Bangladesh to collaborate with scientists around the world to improve the outcome of patients with GBS in Bangladesh and other low- income countries.
Progress Report:
IGOS is fully operational in Bangladesh as of October, 2013. 115 patients have been enrolled in IGOS from Bangladesh and as such our center in Dhaka, Bangladesh, is strongly supporting IGOS as one of the top includers. The 1-year objective in the study proposal was 50 patients, a target that was reached after 9 month. The IGOS web-based data entry support system was used to collect data about demographic factors, preceding infections, rate of progression, neurological deficits, treatment, electrophysiology and other potential prognostic factors in the early phase of disease. Among the 115 GBS patients included from Bangladesh, 77 were male and 38 were female. Biological samples, including blood samples were collected from all GBS patients. All samples were processed and stored at -700 C as per standard protocol. We recorded CSF data (protein, glucose and cell count) from all GBS patients. In addition, we collected and stored 84 CSF samples; 75 of these CSF samples were processed within 2 hrs and stored at -700 C for future proteomics studies. In addition, whole blood samples. were collected for future DNA extraction from all included GBS patients. NCS and EMG study on 93 (81%) patients. First results show that patients can be classified as AMAN (n=49), as AIDP (n=26), and AMSAN (n=7). Two patients had a normal nerve electrophysiology and 1 patient could not be classified. Two patients were excluded from the study as they were finally diagnosed as acute transverse myelitis during follow up. We intend to further include patients and follow-up data for IGOS until the end of the study and will participate in the expertise groups for conducting various IGOS related research projects.
As for Objective two, no procedures have been yet performed. A protocol for the procedure has been developed and will be submitted to the internal IRB in Bangledesh.
ALPHA-1-ANTITRYPSIN AS A NOVEL THERAPY FOR CIDP
Maureen A. Su, MD, Assistant Professor, Department of Pediatrics and Microbiology/Immunology, University of North Carolina at Chapel Hill
Current treatments for CIDP do not work optimally for many patients. Alpha-1-antitrypsin (A1AT), a circulating serine protease inhibitor, re- verses certain autoimmune diseases by exerting potent immunomodulatory effects. Whether A1AT is effective in reversing autoimmune peripheral neuropathy is unknown. It may be able to reverse autoimmune peripheral neuropathy by increasing the frequency of suppressive regulatory T cells (Tregs).
In this proposal, a new CIDP mouse model developed by our group to test whether A1AT might be a potential new treatment option for CIDP. Since A1AT is safe and approved for replacement therapy in humans, findings from this study have the potential for immediate translation to CIDP patients.
We recently reported that female mice with a dominant Autoimmune Regulator (Aire) G228W patient mutation on the autoimmune-prone NOD background (NOD.AireGW/+ mice) spontaneously develop autoimmune peripheral neuropathy between 15 and 22 weeks of age (Su et al. 2008; Su et al. 2012). Additionally, this spontaneous peripheral neuropathy shares multiple features with sporadic CIDP in humans (Su et al. 2012). These similarities suggest that NOD.AireGW/+ mice may be utilized as a CIDP model for p testing of potential immunomodulatory agents to reverse neuropathy.
Female NOD.AireGW/+ mice were treated with a course of A1AT or human albumin (negative control) as described in (Koulmanda et al. 2008). 6-8 week old mice were treated with each reagent at a dose of 2 mg per day every 3rd day for 5 treatments total. Unexpectedly, mice treated with human albumin (negative control) experienced anaphylaxis with the 5th dose, which resulted in a high rate of mortality in the control group. We attempted different dosing schedules and routes of administration to prevent albumin anaphylaxis without success. We therefore switched to vehicle control treatment rather than using albumin as a negative control. With this strategy, we saw a modest decrease in neuropathy-free survival in 6-8 week old mice treated with A1AT compared to vehicle control (Figure 1). No significant differences, however, were seen in histology scores of immune infiltration in sciatic nerves (Figure 2). Furthermore, no differences were seen between A1AT treated and control groups if treatment was given after neuropathy was already present (data not shown). Thus, A1AT as a monotherapy slightly delays the development of neuropathy, but does not reverse already established neuropathy.
Given these findings, our current efforts have been focused on identifying additional reagents that also delay neuropathy development. Such reagents could potentially be tried in combination with A1AT to reverse neuropathy. A1AT is believed to function through inhibiting T cell production of inflammatory cytokines and by upregulating regulatory T cells. We reasoned that reagents that affect distinct pathogenic pathways may be most effective in augmenting the anti-inflammatory effects of A1AT. We therefore sought to identify reagents that may affect distinct immune mediators, such as B cells and macrophages.
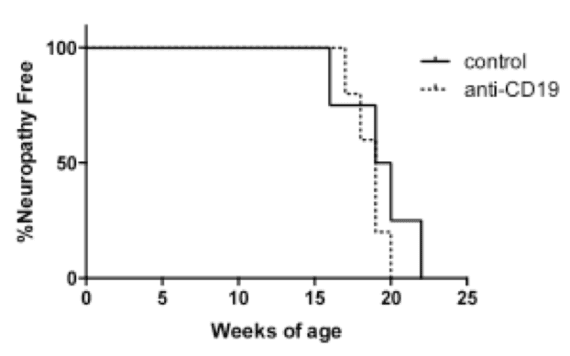
First, we tested whether anti-CD19 antibody, a reagent that downregulates B cells, may protect against neuropathy. No difference was seen in neuropathy-free survival in NOD.AireGW/+ mice treated with either anti-CD-19 antibody or control treatment (Fig 3).
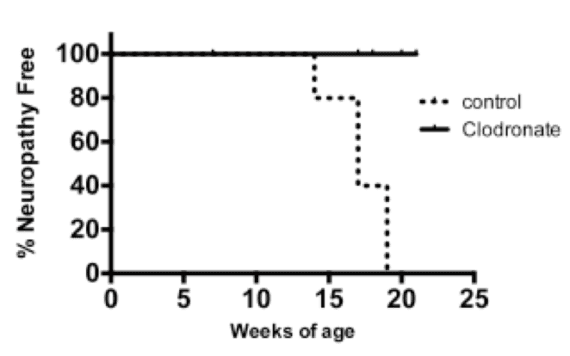
Second, we tested whether clodronate, a liposomal reagent that depletes phagocytic cells such as macrophages and dendritic cells (Calderon et al. 2006), would protect against autoimmune peripheral neuropathy development. Clodronate liposomes prolonged neuropathy-free survival when given to mice at 6-8 weeks of age (Figure 4). Thus, depletion of phagocytic cells appears to prevent autoimmune peripheral neuropathy development. Clodronate treatment after onset of neuropathy, however, was not effective in reversing neuropathy (data not shown).
Future directions: Our ultimate goal is to develop a new strategy for treating patients diagnosed with CIDP. To date, both alpha-1 antitrypsin and clodronate liposome treatment appear to prevent neuropathy before disease onset, but do not have efficacy in reversing (treating) disease. Given the distinct mechanisms by which alpha-1-antitrypsin and clodronate function to dampen autoimmunity, we hypothesize that alpha-1 antitrypsin and clodronate liposome treatment may function in combination to reverse autoimmune peripheral neuropathy. We plan to test this hypothesis in the next stages of this project. We plan to write up our findings in the next year and will utilize this data generated with funding from the GBS/CIDP Foundation to apply for an NIH NINDS R01 grant.
Project Title: LENTIVIRUS TRANSDUCED DENDRITIC CELLS EXPRESSING VIP FOR THE TREATMENT OF CIDP
Jerry R Mendell, MD
Curran-Peters Endowed Chair in Pediatric Research
Professor of Pediatrics and Neurology, Children’s Hospital
Columbus, OH 43205
Background
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is an immune-mediated demyelinating disorder. 35-40 % CIDP patients do not respond well to conventional treatments [intravenous immunoglobulin (IVIg), plasma exchange and corticosteroids] and require an on-going multi-drug regimen. In this study, we used spontaneous autoimmune peripheral polyneuropathy (SAPP) model in B7-2 knockout non-obese diabetic (NOD) mice as an animal model for CIDP which mimics a progressive and unremitting course of CIDP. Auto-reactive T-cells and auto-antibodies directed against myelin protein zero (P0) were detected in SAPP mice. Tolerogenic dendritic cells (tDCs) were shown to home to inflamed and lymphoid tissues to suppress autoimmune responses by reducing the inflammation and priming naïve T cells to be regulatory cells (Tregs). Vasoactive intestinal peptide (VIP) plays an active role in generation of tDCs from bone marrow cells. VIP expressing DCs, transduced with lentivirus vectors (LV-VIP-DCs) exerted a sustained clinical effect on EAE model. Specific aims; We hypostasized that systemic administration of LV-VIP-DCs will suppress immunogenic responses against the myelin protein P0 and prevent nerve fiber demyelination in SAPP mice. Our specific aims included:
Aim 1. SAPP mice will be treated at week 16 with LV VIP DCs to suppress inflammation in the nerve and generate “myelin P0 antigen specific Tregs” to inhibit auto-reactive T cell responses directed against myelin. The rationale for treatment at 16 weeks is to provide a disease modifying effect.
Aim 2. SAPP mice will be treated at week 20 with LV VIP DCs. These mice will have an easily identified clinical neuropathy as well as an inflammatory process in the nerve. The goal is reversal or rescue of the ongoing neuropathy
Results:Generation of LV-VIP-DC
We have successfully generated LV-VIP-DCs in vitro and fully characterized the cells before injections. The results revealed that LV-VIP-DC had tolerogenic capacity. Therapeutic Effect of LV-VIP-DCs in SAPP
Bone marrow derived DCs were transduced to express VIP using a lentiviral vector (LV-VIP). These transduced DCs (LV-VIP-DCs) were then injected intravenously into 16 (before disease onset=AIM 1) and 21 week-old* (after disease onset=AIM2) SAPP mice in order to prevent or attenuate the disease. Outcome measures included behavioral tests, clinical and histological scoring, electrophysiology, real-time PCR, flow cytometry analyses and ELISA assays. LV-VIP-DCs were recruited to the inflamed sciatic nerve and reduced the expression of inflammatory cytokines. A single injection of LV-VIP-DC delayed the onset of disease, stabilized and attenuated clinical signs correlating with ameliorated behavioral functions, reduced nerve demyelination and improved nerve conduction. LV-VIP-DC treatment generated antigen specific immunomodulatory effects in favor of tolerogenicity. This proof of principle study suggests that the use DCs expressing VIP would be applicable to progressive cases of CIDP refractory to standard immunosuppressive therapy including agents like glucocorticoids, rituximab, and intravenous immunoglobulins.
*In our proposal, injection time for AIM2 was 20 weeks of age; however all of the mice started showing clinical decline by 21 weeks of age leading us to choose this time point. End point of the study was 28 weeks of age in our proposal; however considering the rapid decline in the non-treated group we reduced the end point to 25 weeks. This time point allowed us to obtain measurable behavioral and immunological parameters to distinguish the treatment effect.
Data Published in the meetings and peer-reviewed journals
- Our the scientific abstract, number 2007, entitled Treatment Of Experimental CIDP Using Lentivirus Transduced Dendritic Cells Expressing VIP, was accepted for dual oral presentation at the American Academy of Neurology 66th Annual Meeting, April 26 to May 3, 2014 at the Pennsylvania Convention Center in Philadelphia, PA.
Abstract can be reached at: https://www.neurology.org/content/82/10_Supplement/S6.006
(Active link on 6/23/2014 )
- Our abstract entitled “VIP Expressing Dendritic Cell Treatment Ameliorates Spontaneous Autoimmune Peripheral Polyneuropathy” was accepted for a poster presentation at the ASGCT 17th Annual Meeting, May 21-24, 2014 in Washington, DC, USA. This poster presentation was given “Outstanding Poster Award”
- Result of our project was recently published in Molecular Therapy Journal, Molecular Therapyadvance online publication 27 May 2014; doi: 10.1038/mt.2014.77 ***This report acknowledges the support of the GBS-CIDP Foundation for which we are very grateful.
Future Directions
As we continue to move these experiments toward a translational goal, we anticipate that further support for a clinically applicable protocol will come from a repeated LV-VIP-DC infusion paradigm that can be adopted to patients after disease onset. In addition, further studies showing a comparable effect using non-integrating lentiviral vectors will be of value for clinical trials.
Non-Scientific Report
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is an autoimmune disease of the peripheral nerves with clinical features of weakness, sensory loss, imbalance and impaired ambulation that may lead to substantial disability. The need for chronic immunosuppressive treatment is a major treatment challenge in clinical practice. This study described a novel translational approach combining intestinal vasoactive peptide (VIP), lentivirus vector and bone marrow derived dendritic cells (DCs) in the development of a therapeutic tool to treat CIDP. Proof of principle was established in the spontaneous autoimmune peripheral polyneuropathy (SAPP) model of CIDP.
The results showed that allogeneic DCs taken from SAPP mice can be genetically modified to generate tolerogenic DCs and they can be used as a therapeutic tool to treat the SAPP. Single injections of DCs secreting VIP stabilized and attenuated the disease progression in SAPP mice. Treated mice had improved motor functions. Histological analysis showed well preserved myelinated nerve fibers in treated group. We have detected reduced T cell infiltration in the sciatic nerves of treated group compared to the controls. Treatment effect was confirmed by nerve conduction studies showing significantly higher compound motor action potentials as well as conduction velocities. Immunological findings of the study were very encouraging. SAPP mice have a low percentage of regulatory T cells (Tregs=creates long term anti-inflammatory responses) which was also shown in CIDP cases. The treatment increased the regulatory T cells in the lymph nodes and spleen suggesting that VIP expressing DC created immune modulatory effects in favor of tolerogenecity. Although it was a low quantity, we were able to detect injected cells in the inflamed sciatic nerves, lymph nodes and spleen of treated SAPP mice. This data also suggested that increased dose of cells in a single injection or a multiple injection strategy (two or three times) might be more effective to increase the DCs in the target organs and create a robust anti-inflammatory response.
Today, DC therapy is mainly an option for cancer patients. Autologous DCs or DCs obtained from HLA matched donors consists of the major sources of readily available DCs. Use of genetically modified DCs in the treatment of CIDP might require an alternative strategy rather than using integration proficient lentiviral vectors to deliver the therapeutic genes in to DCs. Although it is very minimal, integration proficient lentiviral vectors might bring the risk of insertional mutagenesis in DCs. To avoid this risk we are currently working on testing our integration deficient lentiviral vectors for the treatment of SAPP.
2014 Grant Reports
“PARANODAL AUTOIMMUNITY IN CIDP: DIAGNOSTIC AND THERAPEUTIC VALUE” Dr. Isabel ILLA
Catedràtica Neurologia U.A.B., Unitat Patologia Neuromuscular, Servei Neurologia
Hospital Santa Creu i Sant Pau
A subset of CIDP patients with a clinically distinct presentation have a specific class of antibodies of the IgG4 isotype against proteins at the node of Ranvier, a specialized portion of the nerve between myelinated segments including the paranodeal region. This area of the nerve facilitates impulse propagation. Damage to the region results in weakness and sensory loss. Antibodies to proteins at the paranode are contactin-1 and neurofascin 155 in a limited number of CIDP patients present with distinct clinical features including tremor but do not respond to standard treatment with IVIG or corticosteroids. The immunopathological mechanisms leading to the production of anti-contactin 1 (CNTN1) and anti-neurofascin 155 (NF155) antibodies and to nerve damage in CIDP patients are unknown. The 3 main objectives are as follows.
- First, to expand the number of of CIDP patients with antibodies against paranodal proteins. Regarding this objective we have been able to identify 4 new patients (2 anti-contactin-1 and 2 anti-NF155) positive through alliances with other centers in Spain and Europe. Alliances with other centers will try to confirm the clinical phenotypes described in our original work.
- The second objective aimed to identify specific pathological patterns in CIDP patients’ skin biopsies. The work on this objective has not started yet. We will try to identify more patients with antibodies against paranodal structures and then proceed
- The third objective focused on study of the immunological environment leading to IgG4 anti-CNTN1 – NF155 production, in particular with regard the numbers of regulatory B cells. We have started to set-up the technique for B-reg identification and ran several controls. Once the technique is perfected we’ll start to test neuropathy patients.
Potential outcomes and implications: The confirmation of the association of anti-CNTN1 –NF155 antibodies with specific clinical features in larger cohorts could provide clinically relevant findings for diagnosis. The description of specific immunopathological findings in these patients could identify a subset of patients that would respond B-cell depleting therapies.
GUILLAIN-BARRÉ SYNDROME IN DEVELOPING COUNTRIES AND PARTICIPATION IN IGOSPI: Prof Hubert P Endtz
Co-PI: Prof Deen Mohammad & Dr Bart Jacobs, Site PI: Dr Zhahir Islam
Grant awarded to Erasmus MC, Rotterdam, June 2013
Study agreement ErasmusMC & ICDDR,B, Dhaka, Bangladesh signed October 8, 2013
IRBERB approval ICDDR,B, Dhaka, Bangladesh signed October 10, 2013.
The project involves two studies with the following objectives. (1) Participation in International Guillain-Barré Syndrome (GBS) Outcome Study (IGOS) to define the clinical course and outcome of the Guillain-Barré syndrome (GBS) in developing countries and to identify the preceding infections and other factors that influence outcome, and (2) To investigate the safety of small volume plasma exchange (SVPE) as a new treatment for patients with GBS in Bangladesh and other low-income developing countries where the expense of machines used in Plasma Exchange and the cost of intravenous immune globulin precludes their use.
Objective 1:
IGOS is fully operational in Bangladesh since October, 2013. As of now, 115 patients have been enrolled in IGOS from Bangladesh and as such our center in Dhaka, Bangladesh, is strongly supporting IGOS as one of the top includers. The 1-year objective mentioned the study proposal was 50 patients, a target that was reached after 9 month. This has been a tremendous performance in view of the rapidly deteriorating political situation in the country before and after the 2014 elections that were associated with massive national strikes and disturbances of public life. The dedicated team in Dhaka, however, has managed to keep the inclusion rates high and the target of 50 patients was therefore reached within the time schedule.
The team in Dhaka used the IGOS web-based data entry support system to collect data about demographic factors, preceding infections, rate of progression, neurological deficits, treatment, electrophysiology and other potential prognostic factors in the early phase of disease. Among the 115 GBS patients included from Bangladesh, 77 were male and 38 were female. Biological samples, including blood samples were collected from all GBS patients. All samples were processed and stored at -700 C as per standard protocol. We recorded CSF data (protein, glucose and cell count) from all GBS patients. In addition, we collected and stored 84 CSF samples; 75 of these CSF samples were processed within 2 hrs and stored at -700 C for future proteomics studies. In addition, whole blood samples were collected for future DNA extraction from all included GBS patients. We performed NCS and EMG study on 93 (81%) patients. First results show that patients can be classified as AMAN (n=49), as AIDP (n=26), and AMSAN (n=7). Two patients had a normal nerve electrophysiology and 1 patient could not be classified. Two patients were excluded from the study as they were finally diagnosed as acute transverse myelitis during follow up. We intend to further include patients and follow-up data for IGOS until the end of the study and will participate in the expertise groups for conducting various IGOS related research projects.
Unfortunately, violent country-wide disturbances have started again on January 1, 2015 with a death toll of > 30 as of now, a massive strike of all public transports, closed primary and secondary schools etc. These unfortunate circumstances are reducing access to health services and are thus affecting the number of patients that are reaching the hospitals and our study site ever since January 1, 2015.
Objective 2: Investigate the safety of small volume plasma exchange (SVPE) in patients with GBS in Bangladesh.
A therapeutic study with SVPE, will be set up to determine the feasibility and safety in the treatment of GBS in low-income countries for patients who cannot afford the currently proven effective but very expensive treatment options as e.g. plasma exchange and intravenous immunoglobulins. SVPE is based on the same presumptions as plasma exchange: removal of soluble neurotoxic factors from the blood, including antibodies and complement factors. We developed the protocol on SVPE, and presented a draft version in INC/PNS meeting 2014 held in Dusseldorf, Germany. We took opinion from international experts for this invasive study and adopted in the current protocol. In addition we further improved the protocol in collaboration with experts in infection prevention, intensive care of neurological patients and biostatisticians from Erasmus MC who are experienced in conducting intervention studies in patients with GBS. The protocol for the study has recently been finalized and will now be submitted for local IRB approval. The next two IRB meetings are scheduled in the first week of March and April. We aim to start the first patient inclusions before the summer 2015, pending on the IRB recommendations and approval. Enclosed goes the prefinal study protocol to be submitted soon.
III. DEFINING AND PREDICTING LONG-TERM OUTCOME OF GUILLAIN-BARRÉ SYNDROME IN LOW-INCOME COUNTRIES, PI: Dr. Zhahirul Islam Co-PI: Dr. Bart C. Jacobs, Prof. Quazi D. Mohammed, Prof. Hubert P. Endtz
Grant awarded: ICDDR,B, Dhaka, Bangladesh, July 23, 2014
Study agreement: GBS-CIDP FI & ICDDR,B, Dhaka, Bangladesh , signed October 1, 2014
Progress report:
This Project comprises of the following objectives: (1) To identify new biomarkers for predicting poor outcome in GBS focusing on new preceding infections and anti-neural antibodies, (2) to develop new prognostic models for GBS to predict outcome based on the previously collected data and (3) to develop an outcome measure to define the long-term outcome of GBS in low-income countries.
Objective 1: Biological materials and all clinical data are available from 450 GBS patients that had been previously collected by the ICDDR,B research group to identify new biomarkers. All samples were processed and stored at -700 C as per standard protocol. We started to test sera to define the antecedent infections and antibodies to neural structure. Till date, we tested 600 sera collected from cases and controls as per standard laboratory procedure. We are expecting to analyze the rest of the samples as per schedule. We are also working to develop a database for future analysis of these laboratory data as well as clinical data to identify the intended biomarker; and the team is working with full enthusiasm and tempo maintaining all standard protocols.
Objective 2: As discussed in the proposal, for objective 2 which is to develop new prognostic models for GBS; we are using the available data from the cohort of 450 patients that were enrolled previously. For developing a prognostic model, we are trying to relate early clinical and electrophysiological characteristics and biomarkers to certain clinically relevant endpoints. We have incorporated our data set in statistical software; and we are happy to share that, data cleaning and univariate analysis of relevant variables have already been completed. Our next step is to develop a prognostic model using logistic regression method for which presently the team is preparing work plan. Different factors such as demographic factors, preceding infections, anti-ganglioside antibody response, electrophysiological subtypes and neurological deficits will be considered to develop a simple prognostic model for GBS patients in low-in-come countries.
Objective 3:The third objective of this project is to develop new outcome measures for patients in low-income countries using the International GBS Outcome Study (IGOS) which was launched in Bangladesh. IGOS is fully operational in Bangladesh since October, 2013. We have enrolled 125 patients from Dhaka till date (see above report by Prof Hubert P Endtz). We would like to claim it as a great achievement given the present political turmoil in Bangladesh. We admit that, current political situation has compromised our activities as it limits the patient movement for seeking health care as well as for follow up. Despite of this limitation, the number we reached is still high and our team is continuing to work hard.
Dr. Zhahirul Islam
Dhaka, April, 2015
MOLECULAR MECHANISMS UNDERLYING THE INHIBITORY EFFECT OF GBS-ASSOCIATED ANTI-GANGLIOSIDE ANTIBODIES ON NERVE REPAIR.
Pablo H. H. Lopez, PhD
Twenty percent of GBS patients have a delayed clinical recovery and may have significant disability at the end of a year reflect in part underlying damage to the peripheral nerve axon. The presence of antibodies to sugar containing lipid molecules called gangliosides in myelin membrane are reported to influence the nerve repair. Knowledge of the molecular mechanisms controlling nerve repair could potentialy identify new therapies. Many agents including ganglioside antibodies delay axon regeneration by activation of a common pathway to halt axon regeneration and reinnervation of muscle needed to re-establish motor function in patients.
Aim 1.- To demonstrate the role of collapsing response mediator protein-2 (CRMP-2) as a downstream effector of RhoA/ROCK pathway mediating microtubule destabilization at growth cones (GC) by using a site-specific phosphorylation/inactivation mutant of CRMP-2 at residue T555 (negatively regulated by ROCK).
At present we have initiated the molecular characterization of anti-ganglioside antibody-mediated inhibition of axon regeneration. Preliminary results using time-lapse video microscopy indicates that Dorsal Root Ganglion Sensory neurons nucleoinjected with mutant form of CRMP-2 at residue T555 (mutant CRMP-2-T555A) can overcome neurite retraction induced by anti-ganglioside monoclonal antibody 1B7. We also found increased expression of phosphorylated form of CRMP-2 at residue T555 (inactive form) along the axon shaft prior to growth cone collapse. In order to confirm the specificity of these results, we plan to extent the study to mutant forms of CRMP-2 where key regulatory residues are not related with RhoA signaling pathway.
On the other hand we have setup plasmid concentrations/expression times for insertion by electroporation in living DRG neurons with plasmids containing wild type CRMP-2 or its mutant CRMP-2 T555A in combination with tubulin-GFP. Currently we are studying their effect on the inhibitory activity of mAb 1B7 on axon regeneration in the in vivo model proposed.
Aim 2.- To study the potential therapeutic benefit of modulating the RhoA/ROCK signaling pathway in an in vivo model of axon regeneration using the ROCK inhibitor Y-27632. We experienced some delay in the large scale production of mAb 1B7 (several mg of Ab are required for each animal). For this reason aim 2 will be carry out during this month.
REGULATORY B CELLS IN ANIMAL MODELS OF CIDP, Betty Soliven Neurology Department University of Chicago
, 6030 S Ellis Ave, Chicago , IL, USA
Animal models of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) include but are not limited to a relapsing form of experimental allergic neuritis (EAN), and a spontaneous autoimmune polyneuropathy (SAP) in B7-2 knockout (KO) non-obese diabetic (NOD) mice. We reported that depletion of pathogenic Ab producing B cells and plasmablasts with anti-CD19 mAb limits the severity of SAP when administered prior to the onset of neurological symptoms (such as weakness) but not when given after disease onset. The latter may be due to insufficient time for re- expansion of regulatory B cells (Bregs), which are also partially depleted by anti-CD19 mAb. One of the Breg subsets is the so called IL-10 producing B cells (B10 cells), which comprise 1-3% of mouse splenic lymphocytes and are predominantly found within the CD1d(hi)CD5(+)CD19 subset (refer to as Breg1 hereafter).
Aim: We propose that impaired mechanisms mediated by these Bregs contribute to the persistence or progression of CIDP. This hyposthesis will be examined in SAP, which mimics the progressive form of human CIDP.
The goal of this pilot project is to examine whether B7-2 KO NOD mice exhibit altered numbers or impaired function of Breg1 and B10 cells, and if so, find ways to correct these abnormalities. Amongst the agents to be tested in vitro include granulocyte macrophage colony stimulating factor (GM-CSF) and retinoic acid, both involved in regulating immune tolerance. Flow cytometry, thymidine proliferation, and intracellular cytokine staining will be used in these studies to determine the presence and absence of the Bregs. We will determine if SAP can be suppressed or prevented by infusions of sorted Breg1. Clinical assessment, grip strength measurements, sciatic nerve conduction studies and nerve pathology studies will be performed. These studies will provide insight into mechanisms that regulate the development of CIDP, and may potentially lead to novel therapeutic strategies to increase numbers of Bregs in human CIDP
A series of experiments were performed using the B7-2 knock out (negative) mice that spontaneously develop CIDP and wild type mice that express B7-2 (positive) and do not develop autoimmune neuropathy. The investigators focused on cells that process myelin protein and express them on their cell surface that can then be recognized by lymphocytes and leads to the development of autoimmune neuropathy. A subset of the antigen presenting cells in B 7-2 (negative) mice were defective in presenting myelin protein and these animals did not develop tolerance to the myelin protein leading to a chronic immune response, immune mediated neuropathy with clinical weakness, altered nerve conduction studies and nerve pathology. This effect could be reversed by treatment with a cytokine IL-10 suggesting that measures increasing IL-10 in CIDP patients may modulate disease.
Grant Applications 2015
- Molecular Structural Basis of Autoantibody Recognition in GBS (Kenneth KS. Ng, PhD, University of Calgary, Alberta, Canada)
- Background: Glycolipids are part of cell membranes, and serve as markers or binding sites for molecules such as antibodies. Gangliosides are a type of glycolipid found in the membranes of nerve cells. In GBS, autoantibodies (antibodies against ourselves) attach to gangliosides which in turn impacts neuronal repair mechanisms.
- Goal: to understand how GBS-specific autoantibodies recognize (attach to) glycolipids in a manner that ultimately leads to the development of the disease.
- Methods: Determining the structures of antibody-ganglioside complexes using x-ray crystallography will help researchers understand how GBS-specific antibodies attach to gangliosides. This will allow for the design of antibody-blocking inhibitors (things that could block the binding of antibodies to gangliosides) which will disrupt the progression of GBS and prevent nerve damage.
- Identifying therapeutic targets in GBS models (Rhona McGonigal, PhD; Neuroimmunology Research Group
Institute of Infection, Immunity and Inflammation University of Glasgow; Glasgow, Scotland)- Background: Current treatments for GBS are standard and essentially treat all cases in a similar, non-specific way. However, this ignores the fact that there are different triggers and mechanisms for GBS, and that not every case proceeds in the same way.
- Goal: To create a relevant animal model (mice) that will help us better understand how, when, why, and what causes myelin to degenerate, ultimately revealing potential sites for targeted, specific treatments that will improve patient outcome and reduce the socio-economic burden of the disease.
- Methods: creating an animal model will help to understand the signaling pathway that leads to GBS. This experiment will use genetically modified glycolipid antigen expression (things that trigger autoantibodies to form and attack the body) to study the pathways and signaling related to myelin degeneration and repair. Preliminary data has already indicated that different outcomes are associated with different antigen (antibody triggers) expression, which supports the idea that different cases of GBS could benefit from different therapies and treatments.
- The clinical and functional significance of autoimmune responses to nodal antigens in patients with inflammatory demyelinating neuropathies. (Emily Mathey, Matthew Kiernan, Cindy Lin, Susanna Park, John Pollard and Patricia Armati.; Neuroinflammation Laboratory, Brain & Mind Research Institute, University of Sydney, Australia)
- Background: The myelin coating in neurons is interrupted by small gaps, called nodes of Ranvier, which help propel electrical charges down a nerve. Recent GBS and CIDP research has focused on autoantibodies directed towards these nodes, but it is unclear if these antibodies cause disease or represent a secondary effect unrelated to the emergence of the disease.
- Goal: To examine the significance of antigen (things that trigger antibody formation) targets on these nodes and to correlate them to immunological, neurophysiological, and clinical responses, and to help determine need for IVIg or other therapies.
- Methods: to identify target antigens by developing cell lines required to screen for these antigens. Patients will be followed over 4 years to determine response to treatment and outcome.
- Biobanking for Guillain-Barré syndrome: in search of biomarkers for clinical course and outcome. ( Bart C. Jacobs
Departments of Neurology and Immunology Erasmus MC, University Medical Center Rotterdam, Netherlands)- Background: The International GBS Outcome Study (IGOS) is a prospective cohort study aiming to identify biomarkers to monitor disease activity and predict clinical course and outcome in individual patients with GBS worldwide. Biomaterials are currently collected and stored in >100 centers in 18 countries from at least 1000 patients with GBS followed over 1-3 years.
- Goal: To develop a centralized biobank in order to better research these biomarkers and exploit the full potential of IGOS. From this biobank samples will be distributed to sites all over the world for research projects on biomarkers.
- Methods: Transport DNA and serum samples currently stored at local sites to the central storage facilities in Rotterdam and Glasgow and distribute these biosamples to research centers.
- ICOS, Predictors of Treatment Response (the committee expressed limited interest feeling that monetary support was not required to do this study)
- Background: This is essentially the same study as IGOS, but for CIDP. There is no web-based registry and bio databank for CIDP patients currently.
- Goal: To define the diversity of CIDP and biomarkers of disease activity, and to identify predictors of treatment response and how chronic the disease will be.
- Methods: Using both new onset and established patients, clinical and electrophysiological features will be collected at baseline and during a follow-up period of at least 3 years.
Grant Applications 2016
- Effect of Anti-Contactin-1 Antibodies on DRG Neuron: (Dr. Kathrin Doppler MD, Prof. Dr. Claudia Sommer University Hospital Würzburg)
Synopsis: Antibodies against the paranodal (excitable area of the axon in between the myelinated sections) protein contactin-1 have been reported in a subgroup of patients with CIDP who have tremor and an unsteady gait. Binding of autoantibodies is proposed to lead to impaired nerve conduction. Contactin-1 is also expressed by sensory neurons of the dorsal root ganglia (DRG). It is proposed by the applicant that Ab binding to the DRG causes sensory ataxia (unsteady gait) in patients. Anti-contactin-1 Ab decreases the density of sodium channels on DRG neurons. They therefore hypothesize that binding of anti-contactin-1 to DRG neurons affects the physiology of voltage-gated sodium channels and thus impairs neuronal function. They will determine the effect of anti-contactin-1 Ab on the function of these sodium channels by patch clamp recordings of living cultured mouse DRG neurons after incubation with patients’ or control IgG prepared from Therapeutic plasma exchange material of patients and controls. Ab binding will be confirmed with a cytochrome labeled second antibody.
- Development of a Tolerogenic vaccination for Chronic Inflammatory Demyelinating Polyneuropathy (CIDP): (Cristoforo Comi, MD, PhD. Assistant Professor of Neurology Department of Translational Medicine, University of Piemonte Orientale)
Synopsis: CIDP is an autoimmune disease in which peripheral nerve injury reflects both antibody (Ab) and cell-mediated immune responses against peripheral nerve components or antigens (Ags). Candidate Ags include myelin proteins used to induce experimental autoimmune neuritis (EAN) and nodal/paranodal proteins including contactin-1 and neurofascin. Better knowledge of the spectrum of autoAgs is crucial to develop new specific therapies, such as tolerogenic (or “inverse”) vaccination to turn off the specific immune response. This lab previously developed a vaccine to Experimental Allergic Encephalomyelitis by using nanoparticles encapsulating both auto Ags and immunomodulatory substances that inhibited experimental autoimmune encephalomyelitis
- Loss of Schwann cell plasticity in CIDP: A central role for GM-CSF: (Helmar C. Lehmann, MD Department of Neurology University Hospital of Cologne Kerpener Strasse)
Synopsis: Current available therapies in CIDP are aimed to reduce inflammation in affected nerve fibers. However there is no treatment available to enhance regeneration once nerves are injured. We observed in preliminary experiments that Schwann cells fail to support regeneration of injured nerves when they are exposed to blood components of CIDP patients. Preliminary studies showed a significantly “low expression of cytokine GM-CSF” that might be responsible for the incomplete clinical recovery seen in many CIDP patients. We propose a set of experiments to characterize the pro-regenerative potential of GM-CSF in more detail and to assess GM-CSF as putative biomarker in CIDP. The medium-term goal is to develop a novel treatment strategy to enhance nerve regeneration in CIDP.
- Defining the contribution of B cells to CIDP pathogenesis: (Dr. Kevin O’Connor PhD
Assistant Professor of Neurology, Human and Translational Immunology Program Yale School of Medicine)
Synopsis: Several lines of evidence implicate cellular and humeral factors, either acting independently or in concert, in CIDP pathogenesis. Newly identified proteins in the paranodal region (neurofascin-155, contactin-1 and caspr-1) have recently emerged as antibody targets in about 10% of CIDP patients. However the precise contribution of B cells and autoantibodies to the pathogenic mechanisms remains to be defined. CIDP patients with autoantibodies against these surface paranodal proteins exhibit distinct clinical symptoms; moreover, patients with such antibodies seem to respond to CD20-targeted (B cell elimination) treatment. We propose a pilot study of four CIDP patients positive for autoantibodies recognizing paranodal antigens, four CIDP patients with no detectable levels of these autoantibodies and a group of age-matched healthy controls.
- Analysis of antibody responses against nodal/paranodal antigens in an Austrian cohort of patients with Guillain-Barré Syndrome: (Julia Wanschitz, Department of Neurology, Medical University of Innsbruck, Austria)
Synopsis: Nodal and paranodal proteins have been identified as novel targets for antibody responses in Guillain-Barré Syndrome (GBS). The frequency of these antibodies and their association with distinct subtypes of GBS, however, remain poorly investigated. They will 1) to analyze the frequency of antibody responses against characterized antigens Contactin1, Neurofascin155, and CASPR2 in an Austrian cohort of GBS patients and 2) to identify novel nodal/paranodal antigens in GBS.
- An Italian Multicenter Network for the diagnosis and therapy of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and of its variants Proponent: ( Eduardo Nobile-Orazio Neurology, Department of Medical Biotechnology and Translational Medicine, Milan University
Synopsis: Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a chronic and disabling disease of the peripheral disease of the nervous system that often improve with immune therapy. Several variants have been included under this term but their relation to CIDP is not established considering their sometime different response to therapy. The objective of this study is to determine the frequency and characteristic of these variants, the diagnostic criteria used for their diagnosis, their possible evolution into typical CIDP, the association with specific antibodies, and their response to therapy
Grant Applications 2017
Enhance peripheral nerve repair by modulating macrophage subsets:
(Gang Zhang, PhD Assistant Professor of Neurology University of Texas, Health Sciences Center at Houston)
Synopsis: Autoimmune neuropathies consist of a group of peripheral nerve disorders associated with dysregulation of adaptive and innate immune responses. Among these disorders, Guillain-Barré syndrome (GBS) is one of the most common causes of acute flaccid paralysis worldwide. Two immune modulating therapies, namely, Intravenous immunoglobulin (IVIg) and plasma exchange, have been developed to treat GBS. However, there are many disadvantages including high cost, supply shortages, and multiple side effects that are usually associated with high dose and long infusion time of IVIg. Therefore, new therapeutic strategies that can limit the nerve injury during the acute phase of the disease and enhance repair during recovery period are highly desirable. It is in this context we propose a novel therapeutic strategy of modulating macrophage polarization (promote M2 polarization) for treating GBS. Macrophages are central regulators of inflammation.
Probing the role of skin biopsy in CIDP nodo-paranodopathies:
(Raffaella Lombardi,1 Jerome Devaux,2 Andrea Cortese,3 Giuseppe Lauria1)
Synopsis: CIDP is a heterogeneous pathology that critically lacks of diagnostic and prognostic biomarkers. The recent identification of IgG4 anti-neurofascin 155 (Nfasc155) or anti-contactin 1 (CNTN1) antibodies in a subset of patients has widened the spectrum of CIDP phenotypes. These patients how disabling tremor, poor response to intravenous immunoglobulin, and distal and sensory disturbances. Cell-adhesion molecule Nfasc155 and CNTN1 are expressed on the paranode, and play key roles on sodium channel clustering and glia-axon interactions. Our preliminary results on skin biopsies from three IgG4 Nfasc155-positive CIDP patients revealed complete loss of Nfasc155 staining at the paranodes, asymmetrical paranodes and widening of the nodes of dermal myelinated nerve fibers. One IgG4 CNTN1-positive CIDP patient showed abnormal nodal/paranodal immunostaining with different features as compared with IgG4 Nfasc155-positive patients suggesting specific changes.
Flavivirus and arbovirus associated GBS in South East Asia:
(Umapathi Thirugnanam, Senior Consultant Neurologist, National Neuroscience)
Synopsis: A number of antecedent infections are associated with Guillain-Barré syndrome (GBS). Recently, symptomatic and asymptomatic Zika virus (ZIKV) infection has been shown to trigger GBS. In the Southern hemisphere the burden of flavi and other mosquito-borne arbovirus infections such as dengue, chikungunya and Japanese encephalitis is considerable. Even in a small, developed country like Singapore, dengue infections can exceed 20,000 a year. Symptomatic dengue has been documented to antecede GBS. Recently we reported that asymptomatic dengue could also trigger GBS. In parts of South East Asia, GBS cases increase after rainy season when mosquitos breed and dengue infections are common. The anecdotal experience of doctors working in these region suggests that diarrhea associated GBS is infrequent. The actual burden of dengue and other arbovirus, including ZikV, associated GBS is uncertain. The ZikV strain responsible for the current epidemic is Asian, described in 1960-70s in South East Asia. It is conceivable that ZikV isendemic in this region. The serological response to secondary infections in patients previously infected with dengue or other flavivirus may also impact on the propensity for these viruses to induce dysimmunity. These questions on immunopathology of flavivirus and arbovirus related GBS are particularly pertinent as the search for vaccines is intensified and large scale vaccinations are expected shortly.
